About Me
I am a Computer Science MSc student at Bilkent University, supervised by Assoc. Prof. Ercüment Çiçek. I also work as a Research and Teaching Assistant at Bilkent University, where I previously completed my B.Sc. in Computer Science.
My research centers on deep learning for biomedical data analysis. I am currently focusing on transformer-based models for detecting copy number variations from read-depth signals.
My research interests include:
- Deep Learning
- Bioinformatics
- Explainability
- Privacy
- Generative Models
- CNV Detection
- Genomics
- Transcriptomics
Publications and Preprints
LYCEUM: Learning to call copy number variants on low coverage ancient genomes
Bioinformatics, Volume 41, Issue Supplement_1 (ISMB-ECCB 2025)
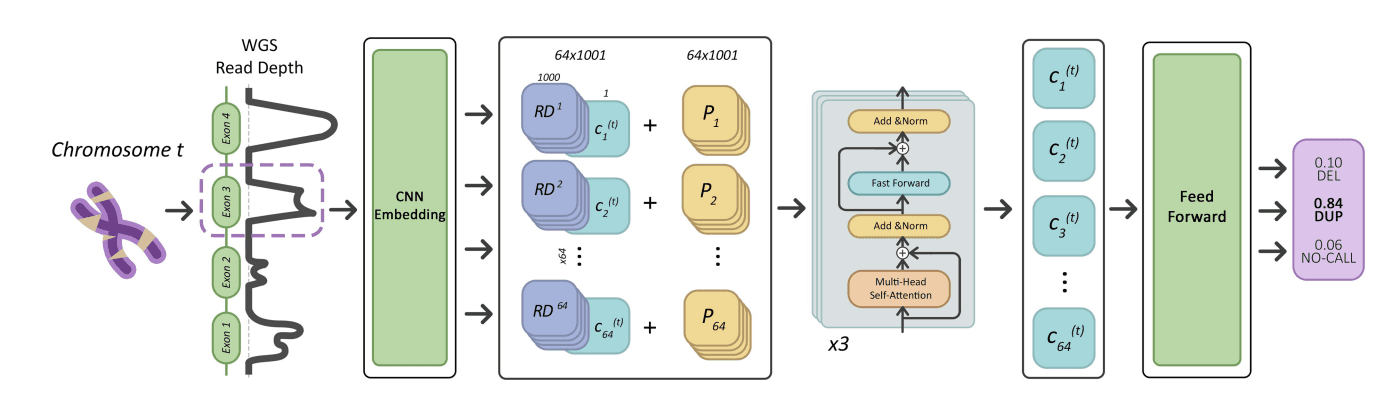
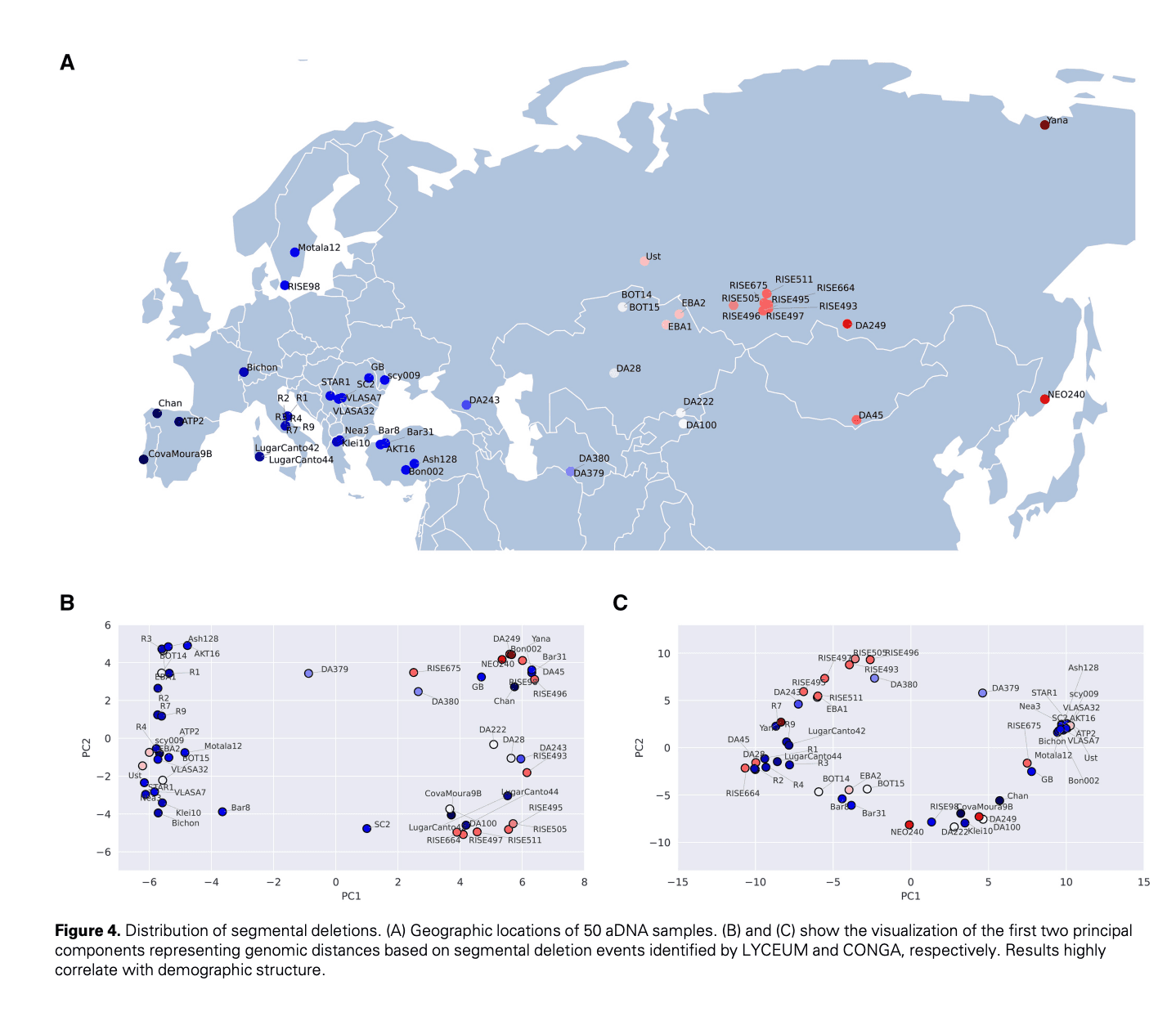
Abstract
Copy number variants (CNVs) are pivotal in driving phenotypic variation that facilitates species adaptation. They are significant contributors to various disorders, making ancient genomes crucial for uncovering the genetic origins of disease susceptibility across populations. However, detecting CNVs in ancient DNA (aDNA) samples poses substantial challenges due to several factors: (i) aDNA is often highly degraded; (ii) contamination from microbial DNA and DNA from closely related species introduce additional noise into sequencing data; and finally, (iii) the typically low coverage of aDNA renders accurate CNV detection particularly difficult. Conventional CNV calling algorithms, which are optimized for high coverage read-depth signals, underperform under such conditions. To address these limitations, we introduce LYCEUM, the first machine learning-based CNV caller for aDNA. To overcome challenges related to data quality and scarcity, we employ a two-step training strategy. First, the model is pre-trained on whole genome sequencing data from the 1000 Genomes Project, teaching it CNV-calling capabilities similar to conventional methods. Next, the model is fine-tuned using high-confidence CNV calls derived from only a few existing high-coverage aDNA samples. During this stage, the model adapts to making CNV calls based on the downsampled read depth signals of the same aDNA samples. LYCEUM achieves accurate detection of CNVs even in typically low-coverage ancient genomes. We also observe that the segmental deletion calls made by LYCEUM show correlation with the demographic history of the samples and exhibit patterns of negative selection inline with natural selection.
ExactCN: Predicting Exact Copy Numbers on Whole Exome Sequencing Data
Submitted to RECOMB 2026; https://www.biorxiv.org/content/10.1101/2025.11.24.690086v1.full.pdf
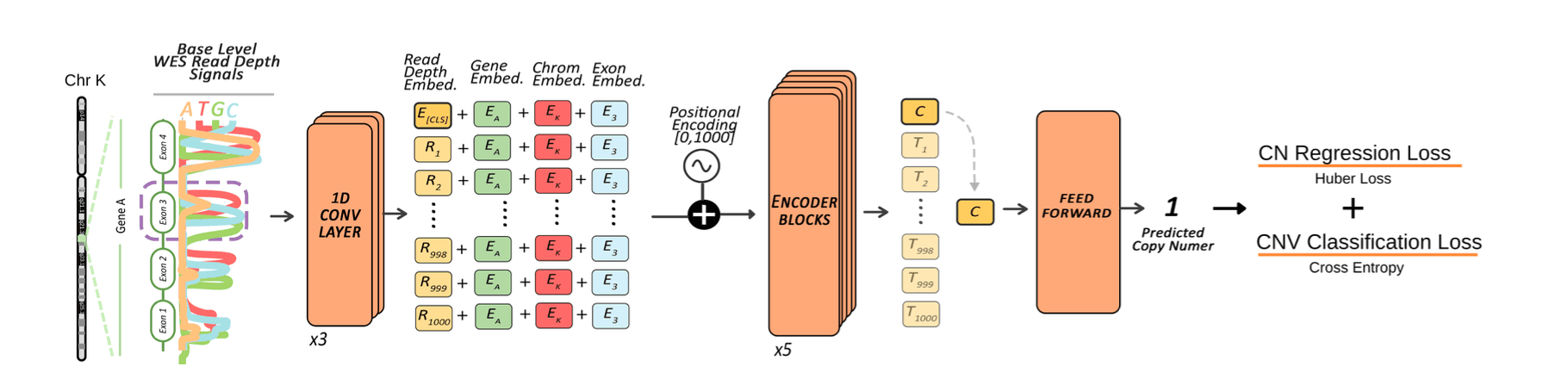
Abstract
The quantification of the precise copy number variations (CNVs) is crucial to understanding the effects of gene dosage, disease severity, and therapeutic response. Although whole-exome sequencing(WES) offers a cost-effective solution for CNV detection in a clinical setting, it introduces several biases, including those related to sequence length, GC content, and the use of targeting probes. Consequently, estimating exact copy numbers remains challenging, especially for WES data. Here, we present ExactCN, a deep learning-based method for estimation of exact copy numbers from WES data per exon. The architecture integrates convolutional layers that extract local read-depth patterns with transformer encoder blocks that capture genomic context and handle sequencing noise. ExactCN is trained on WES samples from the 1000 Genomes Project, using matching WGS-based calls as semi-ground truth. In benchmarks, ExactCN improves the state-of-the-art integer CNV calling performance by reducing the macro-averaged mean absolute error (MAE) from 0.91 to 0.62 and the macro-averaged root mean squared error (RMSE) from 1.31 to 0.78. It also achieves an overall Pearson correlation of 0.669 and Spearman correlation of 0.550, improving the second-best method by 0.641 and 0.482, respectively. Furthermore, a fine-tuned and specialized version of ExactCN for aggregate CNV detection in clinically important duplicated genes SMN1/2 achieved a macro averaged F1-score of 0.657, and mean absolute error of 0.3. These results substantially improves the state-of-the-art performance and demonstrates the model's applicability to both research and clinical genomic analyses.
CHALLENGER: Detecting Copy Number Variants in Challenging Regions Using Whole Genome Sequencing Data
Submitted to RECOMB 2026; https://www.biorxiv.org/content/10.1101/2025.11.23.690083v1.full.pdf
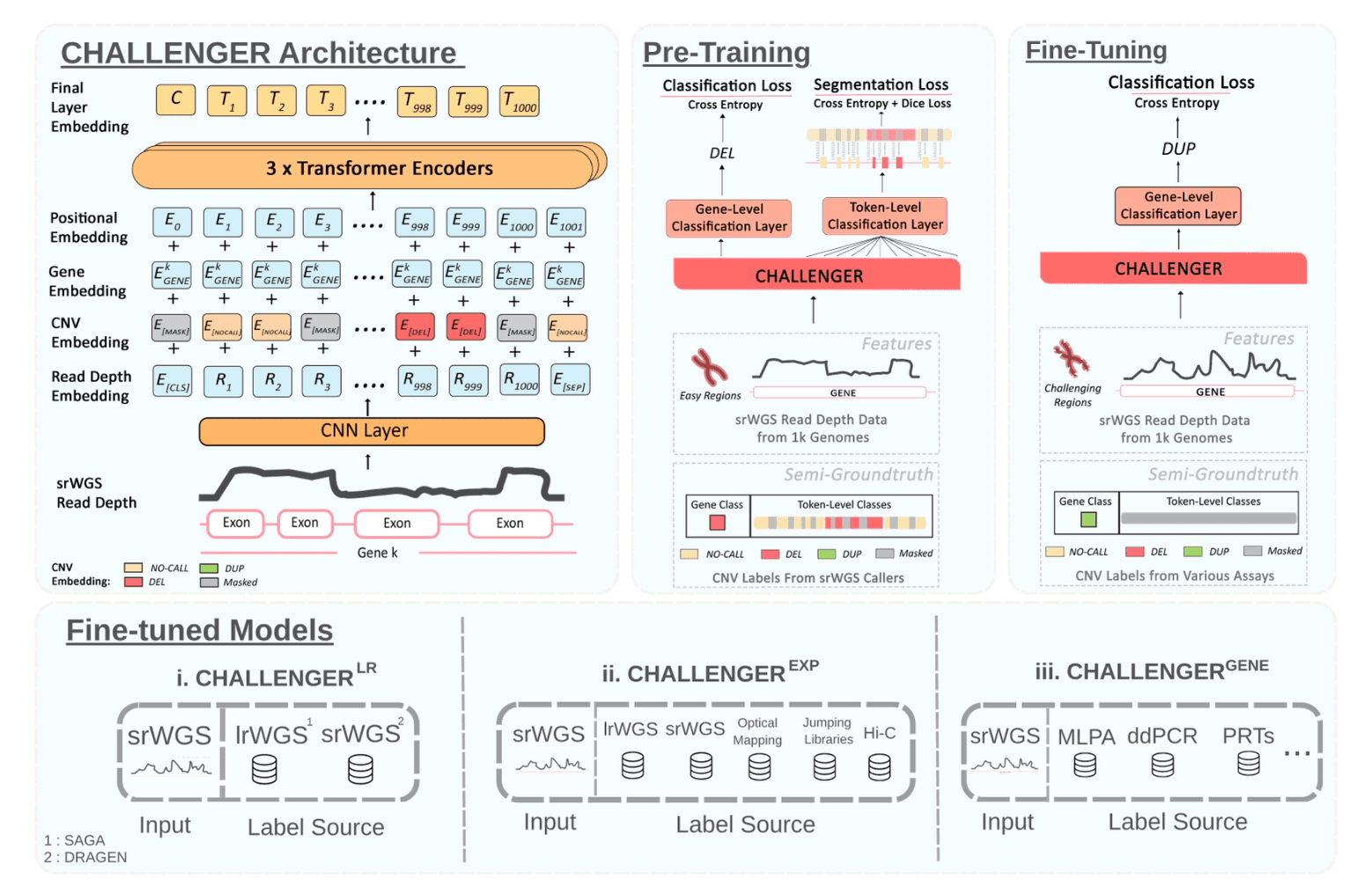
Abstract
Copy number variation (CNV) detection remains a major challenge in whole-genome sequencing (WGS) data, particularly within repetitive, duplicated, and camouflaged genomic regions where short-read sequencing (srWGS) often fails to produce confident alignments. Although long-read WGS (lrWGS) substantially improves structural variant resolution, its high cost limits widespread adoption, especially in clinical settings. To address these limitations, we introduce CHALLENGER, a masked language modeling-based approach for clinical CNV detection using short-read depth signals over coding regions. While the model uses only short-read data as input, it can make calls typically accessible only with long reads, providing a cost-effective way to obtain information characteristic of both technologies. The model is pre-trained on semi-ground truth calls made on srWGS data and then fine-tuned using (i) lrWGS-derived, (ii) human expert-labeled, and (iii) experimentally validated CNV call sets, enabling it to learn technology- and labeling strategy-specific variant signatures hidden within srWGS profiles and to operate in challenging genomic regions. We show that our short-read-only approach improves the state-of-the-art CNV detection F1-score by 40.8%, while, for the first time, capturing 80.3% of CNVs that can only be detected using long reads in challenging genomic regions. The improvement in F1-score in the set of human experts calls is 70.5% for duplications, and 24.6% for deletions in challenging genes. We also specialize CHALLENGER on paralog genes SMN1/2, AMY1/2, and NPY4R, and show that it can improve the performance on experimentally validated call sets while being able to make paralog-specific calls in addition to aggregate calls.
Experiences
Research Assistant
Cicek Lab, Bilkent University | Jan 2024 - Present- Working on transformer-based models for CNV calling using noisy whole-genome depth signals.
- Developing reinforcement-learning-based defense mechanisms for genomic data beacons.
- Contributed to RNA velocity prediction using variational autoencoders.
Research Assistant
Komorowski Lab, Uppsala University (Sweden) | Jul 2023 - Nov 2023- Conducted interpretable ML research on RNA-seq data related to SIV vaccine studies.
- Identified candidate biomarker networks associated with vaccine protection.
iGEM Team Member
Synthetic Biology Research, Bilkent University | Jan 2023 - Jul 2023- Worked on SMA therapeutic strategies by enhancing SMN1 expression.
Machine Learning Research Intern
InfoDif Technology | Jun 2022 - Sep 2022- Implemented ML and computer vision algorithms for defense-related applications.
Teaching Experience
Teaching Assistant at Bilkent University:
- CS464: Introduction to Machine Learning (Spring 2025)
- CS201: Introduction to Data Structures and Algorithm Analysis (Fall 2025)
- CS319: Object Oriented Software Engineering (Fall 2024)
- CS101: Algorithms and Programming I (Fall 2024)
Academic Service
- Reviewer: Research in Computational Molecular Biology (RECOMB) Conference, 2026
- Reviewer: Research in Computational Molecular Biology (RECOMB) Conference, 2025
- Poster Presenter: Google DeepMind MenaML, 2025
- Paper Presenter : UCLA Computational Genomics Summer Institute (CGSI), 2025
- Volunteer: RECOMB Conference, 2023